| 主要科研方向(领域)最新进展,发展趋势、应用前景 |
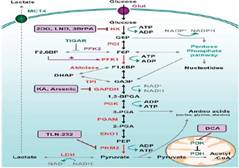
针对beat365中国在线体育目前三个主要科研方向,查阅其国内外发展近况,获知近年来,随着分子生物学、功能基因组学以及蛋白质组学研究的深入,人类对以肿瘤为代表的各种疾病发生发展的分子机制越来越清楚,这为各种疾病的药物治疗提供了一种新的模式,即依据已知疾病发生中涉及的异常分子,包括蛋白或基因,设计和研制针对特定信号通路、蛋白分子或基因靶点的药物,或采用适宜的手段将药物对细胞、组织或器官进行靶向,以选择性地治疗或杀伤异常细胞,减小对正常细胞的损害。当前,多种肿瘤靶向药物已经由实验室走向临床应用,产生了巨大的社会效益,肿瘤的靶向治疗已经成为当前医学研究领域的热点。 我们相信,借鉴肿瘤靶向的成功经验,将靶向手段应用于治疗各种疾病的结构特异性小分子药物研究,将对药学研究者保卫人类健康具有不可估量的意义。 (一)抗菌及细菌多重耐药靶向药物、代谢性疾病新靶点药物研究进展 1.1基于PDF靶标酶分子对接技术的中药抗菌物质筛选研究进展 肽脱甲酰化酶(Peptide deformylase,PDF)是细菌生长过程中蛋白质合成所必需的一个酶。原核生物核糖体介导的蛋白质合成中,在形成起始复合物之前,甲硫氨酸-tRNA先进行甲酰化酶催化的甲酰化,生成甲酰甲硫氨酸-tRNA。在肽链延长过程中,先由PDF催化脱甲酰化,然后由甲硫氨酸氨肽酶(MAP)脱去启动时接上的甲硫氨酸。由PDF催化的细菌新生多肽氮端脱甲酰化是绝大部分细菌正常生长必需的,而在哺乳动物中却没有这个过程。由此,PDF成为新的抗菌药物作用靶点。PDF的活性中心由3个高度保守的基因序列围绕1个金属离子构成。基因组分析表明,PDF同源基因在细菌中广泛分布,由此推测PDF抑制剂类抗菌药物可能会具有广谱抗菌性。 自20世纪90年代末起,PDF抑制剂类抗菌药物成为国内外的一个研究热点。由于PDF作为抑菌靶点的生化过程很明确,其晶体结构和活性部位也比较清楚,其抑制剂类抗菌药物的研究进展较快。目前,British Biotech和GeneSoft的BB-83698,Vicuron的VRC-4887等几个化合物已进入临床研究,它们对金黄色葡萄球菌(包括耐甲氧西林者)、肺炎链球菌、肠球菌和一些耐药链球菌同样有效,因此,PDF抑制剂的研究已经成为了前景乐观的新的抗菌药物研究领域。 目前,多采用化学合成方法获得PDF抑制剂,而从中药材中寻找此类抗菌物质则鲜有报道。天然药物化学成分复杂,为提高化学成分活性评价的效率,可采用近年来已发展日趋成熟并且具有较高预测精度的基于靶点的分子对接技术。 计算机辅助药物设计中的分子对接技术是通过研究小分子配体与受体生物大分子相互作用,预测其结合模式,进而实现基于结构的药物设计方法。根据配体与受体作用的“锁匙原理”,分子对接可以有效地确定与靶受体活性部位空间和电性特征相匹配的小分子化合物,目前广泛应用于数据库搜索及虚拟组合库的设计和筛选研究中。 1.2靶向逆转细菌多重耐药机制研究进展 随着抗生素在临床的广泛使用,细菌的耐药性已引起了研究者的高度重视,20世纪90年代,WHO首次认为细菌对抗生素的耐药问题已经成为了全球性问题。近年来随着广谱抗菌药物的研制开发和广泛应用,多药耐药细菌(MDROs)引起的医院感染在全球更呈上升趋势,尤其在儿童医院,多药耐药细菌感染是引起死亡的重要原因,因此,细菌的多药耐药机制的研究成为了当今科学研究的热点,已经发现的细菌多药耐药机制有: ①细菌产生灭活的抗菌药物酶。使抗菌药物失活是耐药性产生的最重要机制之一,使抗菌药物作用于细菌之前即被酶破坏而失去抗菌作用,这些灭活酶可由质粒和染色体基因表达。 ②抗菌药物作用靶位改变也是细菌耐药的重要机制。由于改变了细胞内膜上与抗生素结合部位的靶蛋白,降低与抗生素的亲和力,使抗生素不能与其结合,导致抗菌失败,如肺炎链球菌对青霉素的高度耐药就是通过此机制产生的,细菌与抗生素接触之后产生一种新的原来敏感菌没有的靶蛋白,使抗生素不能与新的靶蛋白结合而产生高度耐药。靶蛋白数量的增加,即使药物存在时仍有足够量的靶蛋白可以维持细菌的正常功能和形态,导致细菌继续生长、繁殖,从而对抗抗菌药物产生耐药。如肠球菌对β-内酰胺类的耐药性是既产生β-内酰胺酶又增加青霉素结合蛋白的量,同时降低青霉素结合与抗生素的亲和力,形成多重耐药机制。 ③改变细菌外膜通透性也是细菌耐药的机制之一。很多广谱抗菌药都对铜绿假单胞菌无效或作用很弱,主要是抗菌药物不能进入铜绿假单胞菌菌体内,故产生天然耐药,细菌接触抗生素后,可以通过改变通道蛋白性质和数量来降低细菌的膜通透性而产生获得性耐药性。正常情况下细菌外膜的通道蛋白以 OmpF 和 OmpC 组成非特异性跨膜通道,允许抗生素等药物分子进入菌体,细菌多次接触抗生素后,菌株发生突变,产生 OmpF 蛋白的结构基因失活而发生障碍,引起 OmpF 通道蛋白丢失,导致β-内酰胺类、喹诺酮类等药物进入菌体内减少,在铜绿假单胞菌还存在特异的OprD蛋白通道,该通道允许亚胺培南通过进入菌体,而当该蛋白通道丢失时,同样产生特异性耐药。 ④另外,某些细菌能将金土菌体的药物泵出体外,这种泵因需能量,故称主动流出系统,或者外排泵,由于这种主动流出系统的存在及它对抗菌药物选择性的特点,使大肠埃希菌、金黄色葡萄球菌铜绿假单胞菌等对四环素、氟喹诺酮类、大环内酯类、氯霉素、β-内酰胺类产生多重耐药,形成多重耐药状况。 在众多细菌多药耐药机制的研究中,对鲍曼不动杆菌耐药机制的研究起步较晚,原因是该菌在过去10年才逐渐成为重要的医院致病菌;鲍曼不动杆菌的耐药机制研究已引起人们普遍关注,该菌可产生多类β-内酰胺酶,如青霉素酶、头孢菌素酶和金属酶,影响着它们对β-内酰胺类抗生素的敏感性。对喹诺酮药物的耐药与gyrA基因突变有关;对氨基糖苷类耐药则与16S rRNA甲基化酶和药物修饰酶有关。而对多类药物的耐药机制则已证实主要与细菌药物主动外排转运系统相关。如事实上,根据RND泵蛋白的高度同源性,法国Courvalin等于2001年和2004年报道了鲍曼不动杆菌的第一个Resistance- Nodulation-Division (RND)外排系统AdeABC及其双组分调节系统AdeSR,首次证实了鲍曼不动杆菌多重耐药性与RND外排泵的关系。Resistance-Nodulation-Cell Division(RND,耐药-生节-分裂)的概念是1994年李显志等首先以铜绿假单胞菌为例提出的外排泵系统的机制模型。革兰阴性菌RND外排泵系统底物范围较广,每种菌常存在有数个RND系统,但其在耐药性中的作用及其调控机制有所不同,一般仅有1-2种在耐药性形成中起主导作用。苏显中等报道了AdeM泵(属多重药物与毒物外排家族[MATE]); 香港朱耀威等简要地报道了不同鲍曼菌存在的adeABC,adeDE,adeXYZ 基因。但已报道的可能外排泵基因与临床分离菌多重耐药性形成的关系尚未阐明。本研究团队前期实验发现鲍曼不动杆菌所获得的多重耐药菌耐药表型可以用主动外排机制进行解释,尤以AdeABC主动外排系统在鲍曼不动杆菌多重耐药性中起主导作用,且不受已知外排泵抑制剂的作用,使得临床用药陷入困境。鲍曼不动杆菌具有的天然和获得性多重耐药性表型提示该菌的主动外排系统可作为研究靶位。 尽管长期以来国内外学者致力于探索消除或对抗细菌耐药性的有效措施并研制出了一系列抑制剂,如克拉维酸、舒巴坦、CCCP等。但大量的实验研究和临床实践证明这些抑制剂并不能有效地解决细菌的耐药性问题。转变研发新的抗菌药物的策略,寻找新的药物作用靶位势在必行。随着后基因组时代基因表达调控等研究工作的开展,一系列人工干预基因表达的新方法被陆续发现。例如:用反义寡核苷酸/脱氧核酶抑制耐药基因mecR1的表达逆转MRSA耐药性,抑制β-内酰胺酶耐药基因逆转细菌对β-内酰胺类药物耐药性均取得较为可喜的研究结果。研究团队前期在国家自然科学基金资助下, 采用电穿孔法将10-23脱氧核酶导入细菌体内能有效逆转主动外排系统所致的鲍曼不动杆菌多重耐药性。但这种方法不能适用于体内生长的细菌, 探索高效的基因转运系统并将其应用于体内实验以开发有实用价值的新型抗感染性疾病基因治疗药物是迫切需要解决的难题。 耶鲁大学的分子生物学家Ronald Breaher 和他的同事在受到Andrew等人的研究成果启发下发现并命名了核糖开关。核糖开关是mRNA上能够与自由代谢产物或其他小分子配体结合或由于环境条件变化而引起构象变化从而调控基因表达的RNA结构原件。核糖开关多位于受调节基因的5,或3,非翻译区,或者是高等动物基因的内含子区,极少数存在于基因的翻译区,自2002年首次发现维生素B12核糖开关以来,人们先后在细菌、真菌、植物及高等动物代谢相关基因中也发现了核糖开关的存在。核糖开关的配体多是小分子化合物,其中包括很多酶类或辅基,以及这些酶的相应代谢产物。根据配体的类型,迄今发现的核糖开关有十多种,根据核糖开关与配体结合部位二级结构的不同,Montange等将核糖开关分为两大类型:一种是以嘌呤核苷酸核糖开关为代表,其主要特征是配体结合部位是一个单环结构,这种结构使配体诱导的RNA构象变化仅发生在一个窄的区域;另一种以焦磷酸硫胺素(TTP)核糖开关为代表,主要特征是配体结合部位存在两个以上的颈环结构,配体结合后RNA可以在大的区间发生构象变化。核糖开关作为mRNA上能够调控基因表达的RNA结构原件有望成为新型抗生素药物研发的优良靶点。面对细菌耐药性出现的必然性,最终需要新药的研发来解决。 核糖开关作为理想的新型药物靶点具有如下四点基本理由: ①对配体识别的高特异性和基因调控机制的多样性。核糖开关在进化过程中形成了对小分子配体识别的高度特异性,研究数据表明核糖开关的结构性受体比其他任何RNA药物靶点都具有更高的选择性,因此,以核糖开关为靶点的化合物将具有更好的特异性,而对核糖开关受体三维结构的深入研究将能指导化合物的合理设计; ②与宿主代谢的低交叉反应性。除了TPP核糖开关,已知的核糖开关主要位于细菌中,而不是真核生物中。而且真核生物与细菌核糖开关种类、结构具有较大差异,最大限度的降低了细菌核糖开关靶点配体的交叉反应; ③对病原菌代谢调控的有效性。已知核糖开关作用于普遍及必需代谢物和第二信使,这些底物通常与mRNA编码的对于生存或致病性相关蛋白有关。通常,一种核糖开关和其相关特异基因在种系发生上是高度保守的,这使其具有更为重要的生理学意义。 ④既往机制未明的一些抗微生物药物的作用靶点就是核糖开关。如吡啶硫胺(Pyrithiamine)为硫胺(thiamine)类似物,结合TPP核糖开关能够抑制某些细菌和真菌的生长;L-氨乙基半胱氨酸(L-aminoethylcysteine,AEC)和DL-4-还对氧代赖氨酸(DL-4-oxalysine)为赖氨酸类似物,作用于赖氨酸核糖开关抑制某些革兰阳性菌生长;玫瑰黄色素(roseoflavin)为核黄素(riboflavin)类似物,结合FMN核糖开关抑制某些革兰阳性菌生长。 所有直接或间接导致细菌耐药的基因总和称为“耐药组(或抗性组)”(resistome),对耐药组的深入研究,将对预防和战胜细菌耐药问题提供重要帮助,并对于理解细菌耐药相关决定因素在自然生态系统中的功能和生态学作用起到重要作用。由于核糖开关已经进化为特异性结合小分子并通常控制关键代谢通路的表达,随着耐药组序列、结构的进一步阐明,可以预言在耐药组中应能发现相应的核糖开关类型,并可以通过调节核糖开关直接调节单个耐药基因的表达,或者将核糖开关组合于更为复杂的基因环路中作为一种控制元件。目前科研人员正在开展核糖开关合成工程。 细菌耐药基因播撒的关键结构是移动基因元件(mobile genetic elements、MGEs)及相关平台(质粒(plasmids)、转座子(transposon)、整合接合性元件(integrative conjugative elements、ICEs))。在移动基因原件中也可能存在有核糖开关,基于核糖开关机制对移动基因元件的调节,将可阻断移动基因元件所致的水平基因转移(horizontal gene transfer、HGT)。 鉴于以上前景和可行性分析,我们以严重危害术后患者生命的鲍曼不动杆菌为研究对象,系统研究其多重耐药机制以及靶向逆转其耐药的方法,希望可以为有效预防控制多药耐药菌的医院感染提供新的思路。 1.3代谢性疾病新靶点药物研究研究进展 代谢性疾病常常是由各种代谢性危险因素的组合,危害公众身体健康。随着我国人民生活水平的提高,代谢性疾病的患者数量呈逐年增加趋势。尤其是糖尿病及其并发症发病率逐渐升高,在老年人中,其患病率尤其高,且发展迅速。但目前对代谢性疾病包括糖尿病其并发症的治疗效果差。近年来随着新靶点的发现,也促进了相关药物的开发,如葡萄糖激酶(glucokinase,GK)、胰高血糖素(glucogon,GCG)、二肽基肽酶IV(DPP-IV)和蛋白络氨酸磷酸酶-IB(PTP-IB)等新靶点的发现,但至今相关药物还处在研究之中,是目前药物开发的新热点。糖尿病及其肾病的发生及发展是多因素综合作用的结果,认为糖脂代谢紊乱、多种细胞因子、遗传因素及其它如细胞凋亡、炎症细胞浸润、氧化自由基、组织缺氧、血管活性物质等均参与有关,但其中的关键因素并未完全清楚。目前认为内脂素与代谢综合征、肥胖、胰岛素敏感和抵抗以及糖尿病等密切相关,可能成为控制代谢综合征的关键因素,而控制关键因素和靶点就可对代谢综合征,包括糖尿病及其肾病的治疗起到决定性的作用,从而达到良好的治疗效果,因此,对引起其发生的关键因素和新靶点的研究以及相关药物的开发已成为研究的新趋势和热点,也具有非常好的应用前景。 (二)小分子抗肿瘤药物研究研究进展 自从1923年Warburg首次提出针对肿瘤细胞代谢途径研究以来,人们对细胞的糖代谢过程进行了很多的研究。目前已形成了共识的观点:葡萄糖是细胞增殖分化的必要营养物质,它在细胞内通过糖酵解过程释放出丙酮酸,丙酮酸转化为乙酰辅酶A后进入三羧酸循环(TCA),为细胞分化提供必要的能量以及合成细胞膜的基础物质-脂肪酸。此外,葡萄糖还作为碳源通过磷酸戊糖途径参与制造DNA以及RNA。(见Figure1)
Figure1 Figure2 然而,对于肿瘤细胞的生长而言,为了获取维持其快速增殖所需的能量和代谢产物,在代谢过程中的一些关键的酶就发生了突变,尤其更加依赖于对葡萄糖的大量摄取和在缺氧条件下的糖酵解过程(见Figure2)。研究已证实,在糖酵解过程中,一些激酶如己糖激酶(hexokinase, HK)、乳酸脱氢酶A(lactate dehydrogenase A, LDHA)等在各种肿瘤细胞中的活性明显增加;此外,葡萄糖转运酶(Glucose transporter isoforms, GLUT)的活性也明显增加,以此来维持恶性肿瘤细胞增殖的需求。更值得注意的是,最近针对恶性肿瘤细胞糖代谢的研究表明,在恶性肿瘤细胞中糖酵解的限速酶—丙酮酸激酶M2(M2 isoform of pyruvate kinase, PKM2)相对于丙酮酸激酶M1(M1 isoform of pyruvate kinase, PKM1)有显著的过表达,且该酶仅在高速增殖的细胞中才会表达,它的过表达直接促进了葡萄糖在缺氧条件下的酵解,进而为肿瘤细胞生长提供能量和必要的代谢产物。此外,对其深入研究还发现,在PKM2过表达的细胞中,磷酸烯丙醇丙酮酸(PKM2的酶解物)被磷酸甘油酸变位酶(Phosphoglycerate mutase, PGAM)磷酸化后,为肿瘤细胞提供了另外一条糖酵解途径(即能量供应途径)。基于以上分析可知,抑制上述酶中的任何一种,都将抑制肿瘤细胞对葡萄糖的摄取或酵解,特别是抑制PKM2,将会达到选择性地抑制恶性肿瘤细胞在缺氧条件下的糖酵解,从而阻断肿瘤细胞维持其快速增殖所需的能量以及下游代谢物质的供应线。 (三)靶向药物及新型递药系统研究进展 利用计算机程序来模拟化合物与靶标之间的相互作用来指导目标化合物的设计和合成,是近年来国外一门新兴的学科——《化学生物学》的研究内容和主要技术手段。随着越来越多重大疾病关键受体结构及活性位点的的揭示,有目的、有针对性的化合物设计和合成大大减少了科研资源的浪费,具有很大的发展空间和潜力。 目前国外该研究方向的主要思路均是从受体的结构入手,拟定化合物的化学结构因素并赋值,利用计算机(分子对接和分子动力学)进行适当运算,模拟抑制剂与受体之间的相互作用,研究新合成的靶向药物的药理作用、构效关系、分子特征以及三维立体结构,辅以Molispiration和OSIRIS property explorer这两个程序进行新化合物的类药性和毒性程序预测,全面系统地研究靶向药物的药效基团,以此为原则来指导活性更好、毒性更低的新型抑制剂的设计与合成。同时,在化合物设计的过程中还应考虑受体的活性位点大小,因此还需运用Spartan计算软件来预测和控制抑制剂的尺寸大小。因此利用化学生物学手段进行的化学合成并不是传统意义上的盲目合成,而是目标明确、有条理的系统合成,大有取代传统化合物合成结构设计的趋势,同时也是筛选高效、低毒靶向药物的重要手段。 近年来,随着生命科学、材料科学及信息科学的迅猛发展,各学科之间相互渗透、相互促进,大大推动了现代药剂学的发展与进步,使药剂学由经验探索阶段步入了科学研究阶段。药物制剂的新技术、新工艺、新辅料、新材料、新设备不断涌现,制剂基础研究不断深入,剂型与制剂设计理论不断完善,生产工艺技术不断发展与进步,创新药物制剂研发已形成四大类释药系统同时蓬勃发展的新局面:①普通释药系统,如片剂、胶囊剂、注射剂等;②缓释给药系统,如延时释放制剂、长效制剂等;③控释给药系统,如零级速度释药或定时、定位释药制剂、智能化自调控系统等;④靶向给药系统,如脂质体、微囊、微球等。 随着全球医药科研的发展,药物新剂型新技术不断取得突破性进展,大量新剂型、新制剂问世极大地推动了药剂学科的发展,药物递送系统的研究在新药创制中已与新化学实体药研发处于同等重要地位。而且,随着开发新化学实体药投入越来越高,成功率越来越低,世界大多数制药巨头公司都面临巨大的新药创新压力。与开发新化学实体药相比,创新制剂能够提高产品的竞争优势,通过对已有药品的制剂创新和产品改进,如减小药品不良反应、增加新适应症和改善药物疗效、安全性、方便性和顺应性等,可显著改变药物的临床价值和市场价值。 自二十世纪中后期至今,新型释药系统发展迅速,已有35种以上的新释药系统上市,包括缓释给药系统、控释给药系统、靶向给药系统等,如口服缓释小丸、口服渗透泵片、口腔颌鼻腔吸入剂、透皮贴片和贮库型长效注射剂等。2003年美国FDA共批准了35个新药,其中包括21个新分子实体和14个生物技术产品和药物制剂。药物制剂领域已走出制药业的非主流阴影,全球药物创新制剂研究成为制药行业发展最快的领域之一,药物递送技术开发已走在了药物创新的最前沿。
|